AIRRSHIP - Adaptive Immune Receptor Repertoire Simulation of Human Immunoglobulin Production
AIRRSHIP simulates B cell receptor (BCR) sequences for use in benchmarking applications where BCR sequences of known origin are required.
AIRRSHIP replicates the VDJ recombination process from locus creation through to somatic hypermutation. Recombination metrics are derived from a range of experimental sequences allowing faithful replication of true repertoires. Users may also control a wide range of parameters that influence allele usage, junctional diversity and somatic hypermutation rates. The current model extends to human, heavy chain sequences only.
Overview
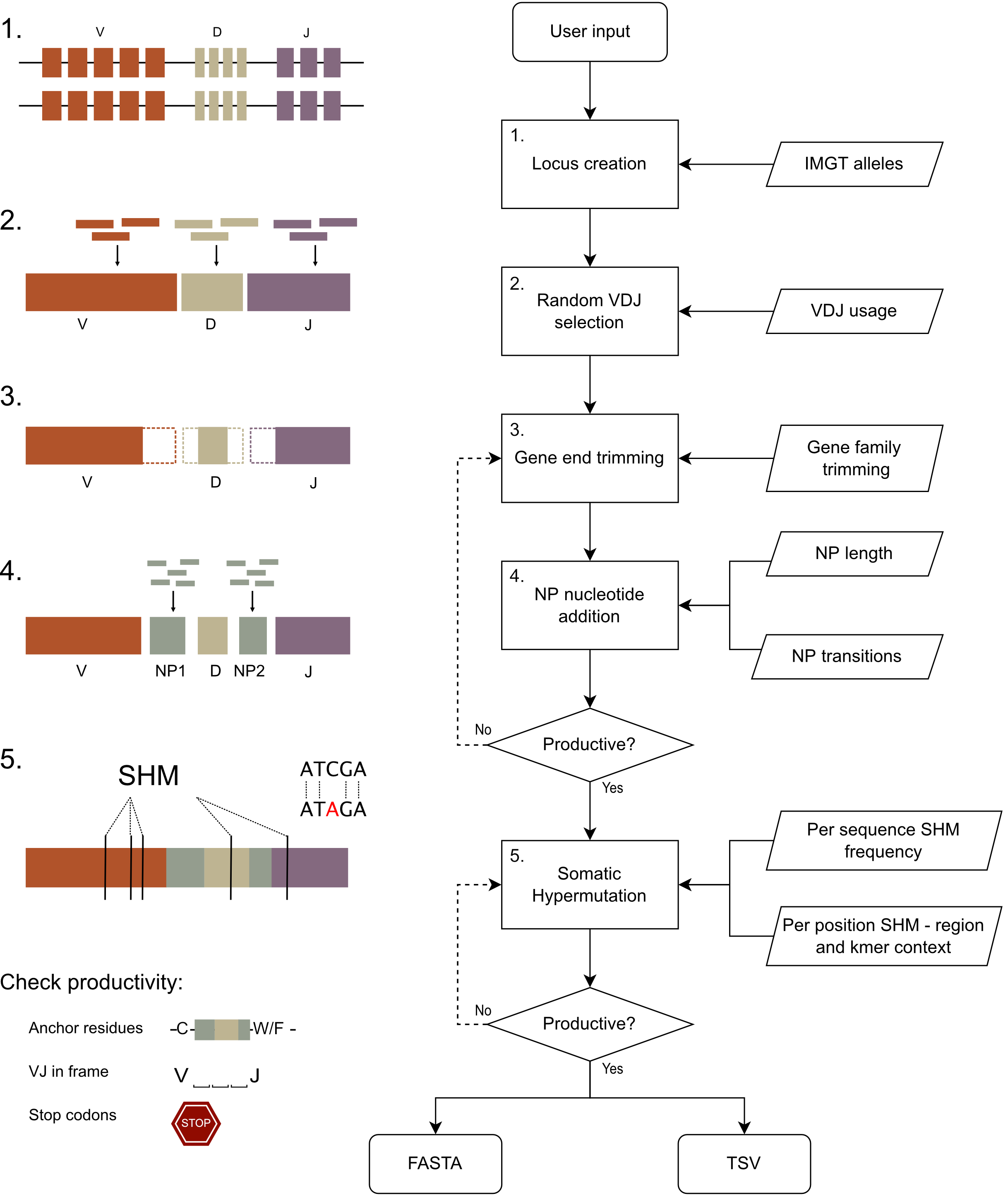
Above: AIRRSHIP simulates human heavy chain BCR sequences, closely replicating experimental data at each step of the synthetic recombination process. Key parameters such as VDJ usage, gene trimming, junctional insertions and somatic hypermutation can all be modified by the user. The FASTA output can then analysed using tools of interest and results easily compared to the tab separated values (TSV) file which acts as a record of the recombination process for each sequence.
Publications
An application note detailing the development and performance of AIRRSHIP is available at Bioinformatics.
If you make use of AIRRSHIP please cite:
Catherine Sutherland and Graeme J M Cowan, AIRRSHIP: simulating human B cell receptor repertoire sequences, Bioinformatics, Volume 39, Issue 6, June 2023, btad365, https://doi.org/10.1093/bioinformatics/btad365
Table of Contents
Quickstart
Download
The latest version of AIRRSHIP can be downloaded from PyPi or GitHub.
Installation
The easiest way to install is using pip, either directly:
pip install airrship
Or after downloading the latest release:
pip install airrship-x.y.z.tar.gz
Requirements
AIRRSHIP intentionally uses only Python standard libraries and requires only the installation of base Python (version 3.7 or above).
Examples
A very small example repertoire is held at the AIRRSHIP GitHub repository to provide an example of the expected output. Larger example repertoire files are available at Zenodo.
Running from the command line
The most basic call to AIRRSHIP requires only an output name.
airrship -o my_repertoire
This will create a repertoire of 1000 unmutated human, heavy chain BCR sequences with metrics derived from experimental distributions.
Four output files will be generated:
- my_repertoire.fasta - final sequences in FASTA format
- my_repertoire.tsv - information regarding sequence generation
- my_repertoire_locus.csv - the simulated locus
- my_repertoire_summary.txt - summary of input commands
Please see Output Files for further details on output file format.
Customising repertoire generation
By default, AIRRSHIP attempts to replicate real experimental repertoires as closely as possible. However, there a large number of command line options that can be used to produce repertoires with specific desired features.
For example, we could create a repertoire with:
- 16,000 sequences
- a locus where as many genes as possible are heterozygous (can't achieve it for every position as not all genes have two alleles)
- balanced usage of the gene families
- no trimming of the 5' end of the D gene
- no insertion of nucleotides between the D and J gene (no NP2 regions)
- include non-productive sequences in output and limit them to 10% of the repertoire
airrship -o complex_repertoire \
-n 16000 \
--het 1 1 1 \
--flat_vdj family \
--no_trim_d5 \
--no_np2 \
--non_productive \
--prop_non_productive 0.1
Full details can be found in Command line Usage.
Note
Occasionally AIRRSHIP may fail to generate a productive sequence from a specific combination of alleles and will print a warning. This should not be of concern unless it happens with high frequency. In this case you may need to check your chosen parameters or input data.
Adding somatic hypermutation
The --shm flag will generate SHM according to observed distributions (see Simulation Model for more information).
airrship -o shm_repertoire --shm
Mutation rates can be controlled by passing a multiplication factor with --shm_multiplier. For example, the below command will create a repertoire with sequences mutated to rates half that as specified in the reference files.
airrship -o shm_repertoire --shm --shm_multiplier 0.5
To request a constant mutation frequency across all sequences, the --shm_flat option can be used. The desired mutation rate or number can be specified with either --mut_rate or mut_num.
The below command will create 1000 sequences, each of which with a mutation rate of 0.08 (i.e. number of mutations in sequence / length of sequence = 0.08). The distribution will be as close to flat as is possible but may fluctuate slightly.
airrship -o shm_flat_repertoire --shm_flat --mutation_rate 0.08
The default SHM algorithm treats each base in the sequence differently, depending on the 5mer context of the base and the region of the sequence it is found in. To make per base mutation independent of sequence context, --shm_random can be used.
airrship -o shm_random_repertoire --shm_random
The per sequence mutation rate will still follow the observed experimental distribution unless --shm_flat is also specified.
Note
Setting mutation rates higher than 0.2 will result in a warning and, depending on the other options specified, may result in very slow performance or a failure to generate sufficient sequences. Other distributions may also be skewed. Mutation rates above 0.5 are not supported.
Using the package in Python
If desired, instead of running from the command line, the package can be imported and a call to main() made within Python, specifying the same parameters as discussed above.
from airrship import create_repertoire
create_repertoire.main(['-o', 'my_repertoire', '--outdir', 'output'])
create_repertoire.main(('-o my_repertoire --outdir output').split())
It is also possible to use individual functions from the package. A simple three step workflow to generate sequences is described below.
Read in data to be used
from airrship import create_repertoire
data_dict = create_repertoire.load_data()
Create a locus from which to generate sequences
locus = create_repertoire.get_genotype()
Generate sequences
sequence = create_repertoire.generate_sequence(locus, data_dict, mutate = True)
generate_sequence returns an individual Sequence class object with all information about its generation stored as attributes. For example, the final mutated sequence can be accessed using sequence.mutated_seq. For full details, see Python.
Simulation Model
1. Locus creation
AIRRSHIP takes IMGT FASTA files of VDJ alleles as input. These files are used to populate artificial heavy chain loci, each including single alleles representing every V, D and J gene. If more than two alleles exist for that gene, the allele included on each chromosome may differ (resembling heterozygous carriage) and the user can control the proportion of genes for which this is desired. By default, this means no one repertoire will contain sequences formed from more than two alleles of a single gene. However, the user can override this requirement and all alleles present in the input dataset will be used (using --all_alleles). The proportion of heterozygous positions can be controlled by the --het argument.
It is possible to override this and use all alleles present in the input dataset (specify with --all_alleles).
2. VDJ recombination
To generate the initial recombined sequence, VDJ alleles are chosen based on gene usage distributions established from published datasets produced by 5’ RACE amplification. Gene segments are recombined from the same synthetic loci only.
VDJ usage may also be requested to be flat at either the gene or family level (--flat_vdj gene or --flat_vdj family).
3. Trimming of gene ends
The 3’ end of the V gene, 5’ and 3’ end of the D gene and 5’ end of the J gene are then trimmed. For each gene end, the number of nucleotides to be removed is sampled from distributions of trimming lengths for each IMGT gene family. To maintain sequence productivity, trimming lengths that would extend past the C-104 anchor in the V gene or the W/F - 118 anchor in the J gene, or that would result in complete deletion of the D gene, are resampled.
Trimming may be turned off at any or all of these gene ends (--no_trim, --no_trim_v3, --no_trim_d5, --no_trim_d3 or --no_trim_j5).
4. Addition of NP nucleotides
AIRRSHIP does not distinguish between N and P nucleotides when insertions are modelled at the VD (NP1) and DJ (NP2) junctions. For each insertion, the number of nucleotides to be added is sampled from distributions of NP1 or NP2 lengths. The first nucleotide to be inserted is then randomly selected with probabilities for each base determined from public data for NP1 and NP2. Addition of further nucleotides follows a position dependent Markov process. For each position in the NP region, the next base is chosen with likelihood determined by a transition matrix that considers the current position in the sequence and the nucleotide base present at that position. When simulating non-mutated sequences and hypermutated sequences, a transition matrix determined from IgD/IgM sequences and from IgA/IgG sequences respectively, is used. This compensates for the inability to determine which positions within an NP region have been hypermutated.
NP addition may be turned off at either or both of the junctions (--no_np, --no_np1 or --no_np2).
5. Somatic hypermutation (SHM)
Somatic hypermutation (SHM) is replicated at both the per sequence and per nucleotide position level. For each sequence, the overall mutation frequency is chosen from a distribution established from published datasets produced by 5’ RACE amplification methods. Each 5mer within the sequence, excluding those where the centre base is an NP nucleotide, is then randomly iterated over. A new base is chosen to replace the centre base of this 5mer, sampled from a set of distributions that give the frequency of each nucleotide occurring at the centre position of each unique kmer for each sequence region (e.g. FWR1, CDR1). If the base chosen differs from the germline base, a mutation is introduced at that position and the process repeats until the number of mutations required to give the desired mutation frequency is reached.
A flat per sequence mutation rate may also be requested (using --shm_flat and specified by either --mut_num or --mut_rate). Each sequence in the repertoire will have the same number or frequency of mutations.
When --shm_random is specified, per position mutation is not explicitly modelled. Instead, each base is mutated randomly, irrespective of it's regional or kmer context. This may be combined with --shm_flat.
6. Removal of non-productive sequences
AIRRSHIP aims to produce only productive BCR sequences, defined here as those where the V and J segments are in-frame, no stop codon is present and the correct junction anchor residues (C-104 and W/F-118) are present. Alleles which do not have the correct anchor residues are excluded when processing the input data. Checks for productivity occur following trimming and NP addition. As certain VDJ combinations may be more likely to result in non-productive rearrangements, multiple attempts are made at trimming and nucleotide insertion using the same allele set to maintain VDJ usage distributions. SHM also commonly renders sequences non-productive, so multiple attempts at introducing hypermutation at the same rate will be made to ensure the per-sequence hypermutation reflects published datasets. All non-productive sequences generated can be included in output by using the --non_productive flag. The proportion of the final repertoire consisting of non-productive sequences can be controlled by using -prop_non_productive.
Commandline Usage
airrship [-h] [-v] -o OUT_NAME [--outdir OUTDIR] [--datadir DATADIR]
[-n NUMBER_SEQS] [--het PROP PROP PROP] [--shm] [--shm_multiplier SHM_MULTIPLIER]
[--shm_flat] [--mut_rate MUT_RATE | --mut_num MUT_NUM] [--shm_random]
[--all_alleles] [--locus LOCUS FILE]
[--flat_vdj {gene,family,False}] [--no_trim]
[--no_trim_v3] [--no_trim_d3] [--no_trim_d5] [--no_trim_j5]
[--no_np][--no_np1] [--no_np2] [--non_productive]
[--prop_non_productive PROP] [--seed SEED] [--species SPECIES]
Parameters
| Option | Details |
|---|---|
| -o, --outname <out_prefix> | Name for repertoire files. Only required parameter. |
| --outdir <out_dir> | Output directory. The current working directory is used if not specified. |
| --datadir <data_dir> | Alternative input data directory. Defaults to airrship/data. Data must be formatted as in the airrship data directory |
| -n, --number_seqs <n_seqs> | Number of sequences to simulate. Defaults to 1000. |
| --het < prop prop prop > | Proportion of genes to be heterozygous, specify as V D J. Values must be between 0 and 1. Not compatible with --all_alleles. Not all genes have more than one allele. The proportion achieved may therefore be lower than requested. Defaults to 0 0 0 (the maximum possible proportion of heterozygous genes using the included IMGT alleles). |
| --shm | Hypermutate sequences according to experimental parameters. Each base will be mutated according to its 5mer context and the mutation frequency for each sequence will match observed distributions. If not specified, sequences will not be mutated. Mutation rates can be controlled by replacing the mut_freq_per_seq_per_family.csv reference file or specifying a --shm_multiplier. |
| --shm_multiplier | Multiplication factor to use on per sequence mutation rate distribution. Defaults to 1, i.e. replicates frequencies from the mut_freq_per_seq_per_family.csv reference file. |
| --shm_flat | Mutate each sequence to the same degree (i.e. return a flat per sequence mutation distribution). Specify degree of mutation using --mut_num or --mut_rate. Will default to a mutation rate of 0.05. |
| --shm_random | Do not mutate individual bases according to kmer context. Each base will have an equal chance of being mutated. |
| --mut_rate <mut_rate> | Mutation frequency for flat SHM only. Value between 0 and 0.6. Not compatible with --mut_num. Defaults to 0.05. |
| --mut_num <number_muts> | Number of mutations for flat SHM only. Not compatible with --mut_rate. |
| --all_alleles | Use all available alleles from all available genes, i.e., do not generate a synthetic 'haplotype'. Not compatible with --het. |
| --locus <locus_file> | Do not generate a new locus, instead specify path to an existing csv file to use as locus for repertoire generation. |
| --vdj_flat {gene, family} | Do not use experimental data to bias VDJ usage, instead use all genes or families evenly. |
| --no_trim | Don't trim any end of any VDJ genes during recombination. |
| --no_trim_v3 | Don't trim 3' end of V genes during recombination. |
| --no_trim_d5 | Don't trim 5' end of D genes during recombination. |
| --no_trim_d3 | Don't trim 3' end of D genes during recombination. |
| --no_trim_j5 | Don't trim 5' end of J genes during recombination. |
| --no_np | Don't insert nucleotides at either gene junction, i.e., do not create NP regions. |
| --no_np1 | Don't insert nucleotides at the VD junction, i.e., do not create NP1 regions. |
| --no_np2 | Don't insert nucleotides at the DJ junction, i.e., do not create NP2 regions. |
| --non_productive | Include non-productive sequences in the output. This includes sequences with out of frame V and J segments, stop codons and/or missing junction anchor residues (C-104 and W/F-118). The majority of sequences produced will be non-productive (~75% using defaults without SHM, ~85% with SHM). Specify --prop_non_productive to control this proportion. |
| --prop_non_productive <prop> | Proportion of sequences to be non-productive. Value between 0 and 1. Use with --non_productive. |
| --seed <seed> | Set random seed. |
| --species <species> | Specify if simulating non-human sequences. Will be used to find the imgt_{species}_IGH[V/D/J].fasta files in the specified --datadir. Deafult is human. |
Python Usage
Overview
For a basic example of importing AIRRSHIP as a package, see here.
Detailed Function and Class Documentation
create_repertoire.load_data
Loads and processes required data files from data folder.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
data_folder |
path
|
Path to data folder with required data. If not specified then uses inbuilt package data. Defaults to None. |
None
|
mutate |
bool
|
Whether to read in data for mutated sequences or not. Defaults to False. |
False
|
Returns:
| Name | Type | Description |
|---|---|---|
data_dict |
dict
|
Dictionary containing all required data for generating sequences. Includes family_use_dict, gene_use_dict, trim_dicts, NP_transitions, NP_first_bases, NP_lengths, mut_rate_per_seq and kmer_dicts. |
create_repertoire.get_genotype
Wrapper that generates a locus for use in sequence generations
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
data_folder |
path
|
Path to data folder with required data. When not specified will use package data. Defaults to None. |
None
|
het_list |
list
|
Proportion of genes [V, D, J] to be heterozygous. Defaults to [1, 1, 1]. |
[1, 1, 1]
|
haplotype |
bool
|
True when only two alleles per gene are to be used. Defaults to True. |
True
|
locus |
path
|
Path to file with predefined locus. Defaults to None. |
None
|
Returns:
| Name | Type | Description |
|---|---|---|
locus |
list
|
List of two dictionaries. Each is a dictionary containing the gene segment as keys and the chosen alleles as values. Format is {Segment : [Allele, Allele ...], ...} |
create_repertoire.generate_sequence
Wrapper to bring together entire sequence generation process.
Recombines, trims and mutates. Optional produces functional sequences (sequences with an in-frame V and J gene, no stop codons and the expected junction anchor residues) or non-functional sequences.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
locus |
list
|
List of two dictionaries. Each is a dictionary containing the gene segment as keys and the chosen alleles as values. Format is {Segment : [Allele, Allele ...], ...} |
required |
data_dict |
dict
|
Output of load_data(). Includes family_use_dict, gene_use_dict, trim_dicts, NP_transitions, NP_first_bases, NP_lengths, mut_rate_per_seq and kmer_dicts. |
required |
mutate |
bool
|
True if SHM to be introduced. Defaults to False. |
False
|
flat_usage |
optional
|
gene, family or False. Gene or family specify that sequences should use all genes or gene families evenly. If false, usage follows experimental distributions. Defaults to False. |
False
|
no_trim_list |
tuple
|
List of 5 Booleans, specifying whether to not trim [all_ends, v_3_end, d_5_end, d_3_end, j_5_end]. Defaults to (False, False, False, False, False). |
(False, False, False, False, False)
|
no_np_list |
tuple
|
List of 3 Booleans, specifying whether to not add [both_np, np1, np2]. Defaults to (False, False, False). |
(False, False, False)
|
shm_flat |
bool
|
True if SHM is to be even across all sequences. Defaults to False. |
False
|
shm_random |
bool
|
True if per base mutation is to be random. Defaults to False. |
False
|
mutation_rate |
float
|
Mutation rate to be used rather than choosing from distribution. Defaults to None. |
None
|
mutation_number |
int
|
Number of mutations to be added rather than choosing from distribution. Defaults to None. |
None
|
mut_multiplier |
float
|
Multiplier to be used on mutation rates pulled from distribution. |
1
|
non_functional |
bool
|
Return non-functional sequences. Defaults to False. |
False
|
Returns:
| Name | Type | Description |
|---|---|---|
sequence |
Sequence
|
Final recombined sequence, with trimming, NP region addition and SHM if requested. |
create_repertoire.Sequence
Represents a recombined Ig sequence consisting of V, D and J segments.
Attributes:
| Name | Type | Description |
|---|---|---|
v_allele |
Allele
|
IMGT V gene allele. |
d_allele |
Allele
|
IMGT D gene allele. |
j_allele |
Allele
|
IMGT J gene allele. |
alleles |
list
|
List of IMGT alleles. |
NP1_region |
str
|
NP1 region - between V and D gene. |
NP1_length |
int
|
Length of NP1 region. |
NP2_region |
str
|
NP2 region - between V and D gene. |
NP2_length |
int
|
Length of NP2 region. |
ungapped_seq |
str
|
Ungapped nucleotide sequence. |
gapped_seq |
str
|
Gapped nucleotide sequence. |
mutated_seq |
str
|
Ungapped mutated nucleotide sequence. |
gapped_mutated_seq |
str
|
Ungapped mutated nucleotide sequence. |
mutated_seq |
str
|
Ungapped mutated nucleotide sequence. |
junction |
str
|
Nucleotide sequence of junction region. |
v_seq |
str
|
Nucleotide sequence of V region. |
d_seq |
str
|
Nucleotide sequence of D region. |
j_seq |
str
|
Nucleotide sequence of J region. |
v_seq_start |
int
|
Start position of V region. |
d_seq_start |
int
|
Start position of D region. |
j_seq_start |
int
|
Start position of J region. |
v_seq_end |
int
|
End position of V region. |
d_seq_end |
int
|
End position of D region. |
j_seq_end |
int
|
End position of J region. |
mutations |
str
|
Mutation events. |
mut_count |
int
|
Mutation count. |
mut_freq |
int
|
Mutation frequency. |
functional |
bool
|
Sequence is functional. |
stop |
bool
|
Presence/absence of stop codon. |
anchors |
bool
|
Presence/absence correct junction anchors. |
inframe |
bool
|
VJ is in-frame. |
__init__(v_allele, d_allele, j_allele)
Initialises a Sequence class instance.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
v_allele |
Allele
|
IMGT V gene allele, required. |
required |
d_allele |
Allele
|
IMGT D gene allele, required. |
required |
j_allele |
Allele
|
IMGT J gene allele, required. |
required |
get_junction_length()
Calculates the junction length of the sequence (CDR3 region plus both anchor residues).
Returns:
| Name | Type | Description |
|---|---|---|
junction_length |
int
|
Number of nucleotides in junction (CDR3 + anchors) |
get_nuc_seq(no_trim_list, trim_dicts, no_np_list, NP_lengths, NP_transitions, NP_first_bases, gapped=False)
Creates the recombined nucleotide sequence with trimming and np addition.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
no_trim_list |
list
|
List of 5 Booleans, specifying whether to not trim [all_ends, v_3_end, d_5_end, d_3_end, j_5_end]. |
required |
trim_dicts |
dict
|
A dictionary of dictionaries of trimming length proportions by gene family for each segment (V, D or J). |
required |
no_np_list |
list
|
List of 3 Booleans, specifying whether to not add [both_np, np1, np2]. |
required |
NP_lengths |
dict
|
Dictionary of possible NP region lengths and the proportion of sequences to use them. In the format {NP region length: proportion}. |
required |
NP_transitions |
dict
|
Nested dictionary containing transition matrix of probabilities of moving from one nucleotide (A, C, G, T) to any other for each position in the NP region. |
required |
NP_first_bases |
dict
|
Nested dictionary of the proportion of NP sequences starting with each base for NP1 and NP2. gapped (bool): Specify whether to return sequence with IMGT gaps or not. |
required |
Returns:
| Name | Type | Description |
|---|---|---|
nuc_seq |
str
|
The recombined nucleotide sequence. |
create_repertoire.Allele
Class that represents a V, D or J allele.
Attributes:
| Name | Type | Description |
|---|---|---|
name |
str
|
The IMGT name of the allele |
gapped_seq |
str
|
The IMGT gapped germline nucleotide sequence |
length |
str
|
IMGT defined length of the allele |
ungapped_sq |
str
|
Ungapped germline nucleotide sequence |
trim_5 |
int
|
Number of nucleotides to be trimmed from 5' end |
trim_3 |
int
|
Number of nucleotides to be trimmed from 3' end |
__init__(name, gapped_seq, length)
Initialises an Allele class instance.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
name |
str
|
The IMGT name of the allele |
required |
gapped_seq |
str
|
The IMGT gapped nucleotide sequence |
required |
length |
str
|
IMGT defined length of the allele |
required |
get_trim_length(no_trim_list, trim_dicts)
Chooses trimming lengths for allele.
Adds two class attributes - trim_3, 3' prime trimming value and trim_5, 5' prime trimming value.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
no_trim_list |
list
|
List of 5 Booleans, specifying whether to not trim [all_ends, v_3_end, d_5_end, d_3_end, j_5_end]. |
required |
trim_dicts |
dict
|
A dictionary of dictionaries of trimming length proportions by gene family for each segment (V, D or J). |
required |
Output files
When used at the command line, AIRRSHIP produces four output files per run:
1. Sequence FASTA
outname.fasta
A FASTA file containing the final simulated sequences. In the case of SHM, these will be the mutated sequences. The FASTA headers correspond to the sequence_id column in outname.tsv.
2. Sequence information TSV
outname.tsv
A tab-delimited file containing information about each sequence and its formation. The file format follows the AIRR-C Rearrangement Schema where possible.
The below columns are present regardless of simulation criteria:
| Name | Description |
|---|---|
| sequence_id | Unique sequence identifier. |
| sequence | Final simulated nucleotide sequence. |
| productive | True if sequence is predicted to be productive. |
| stop_codon | True if the sequence contains a stop codon. |
| vj_in_frame | True if the V and J segments are in frame. |
| v_call | V gene with allele. |
| d_call | D gene with allele. |
| j_call | J gene with allele. |
| junction | Junction region nucleotide sequence. CDR3 plus two conserved codons. |
| junction_aa | Junction region amino acid translation. |
| junction_length | Length of the junction region. |
| np1_length | Length of the combined N/P region between the V and D gene. |
| np1 | Nucleotide sequence of the combined N/P region between the V and D gene. |
| np2_length | Length of the combined N/P region between the D and J gene. |
| np2 | Length of the combined N/P region between the D and J gene. |
| v_3_trim | Number of nucleotides trimmed from the 3' end of the V gene. |
| d_5_trim | Number of nucleotides trimmed from the 5' end of the D gene. |
| d_3_trim | Number of nucleotides trimmed from the 3' end of the D gene. |
| j_5_trim | Number of nucleotides trimmed from the 5' end of the J gene. |
| v_sequence | Part of the sequence originating from the V gene. |
| d_sequence | Part of the sequence originating from the D gene. |
| j_sequence | Part of the sequence originating from the J gene. |
| v_sequence_start | Start position of the V gene in the sequence (1-based closed interval). |
| v_sequence_end | End position of the V gene in the sequence (1-based closed interval). |
| d_sequence_start | Start position of the D gene in the sequence (1-based closed interval). |
| d_sequence_end | End position of the D gene in the sequence (1-based closed interval). |
| j_sequence_start | Start position of the J gene in the sequence (1-based closed interval). |
| j_sequence_end | End position of the J gene in the sequence (1-based closed interval). |
Some columns are present only when SHM is not simulated:
| Name | Description |
|---|---|
| gapped_sequence | Simulated nucleotide sequence with gaps inserted according to IMGT schema. |
Some columns are present only when SHM is simulated:
| Name | Description |
|---|---|
| shm_events | Comma-delimited list of mutation events. In the format position:base>mutated_base |
| shm_count | Number of mutations in the sequence. |
| shm_freq | Mutation frequency (number of mutations divided by length of sequence) |
| unmutated_sequence | Unmutated simulated nucleotide sequence. |
| gapped_unmutated_sequence | Unmutated simulated nucleotide sequence with gaps inserted according to IMGT schema. |
| gapped_mutated_sequence | Mutated simulated nucleotide sequence with gaps inserted according to IMGT schema. |
3. Locus file
outname_locus.csv
A two column CSV file containing the alleles chosen for each simulated "chromosome". Can be used in subsequent runs to simulate sequences from the same genetic background.
4. Summary file
outname_summary.txt
A text file listing the arguments provided to the AIRRSHIP call.
Input files
AIRRSHIP uses various input data in order to accurately replicate real repertoires. If desired, an alternative data directory can be specified. Files in this directory must follow the format of the included data, including exactly matching headers and column order, and be named in the same manner.
Scripts for producing input files
A selection of scripts are in included in the scripts folder on Github and can be used to process AIRR TSV files into the required formats for AIRRSHIP input.
Each reference file requires that certain AIRR-C format columns are present - to produce all reference files, the following columns must be included in your input: sequence,sequence_alignment, germline_alignment, fwr1, fwr2, fwr3, cdr1, cdr2, cdr3, v_call, d_call, j_call, np1_length, np2_length, v_sequence_end, d_sequence_start, j_sequence_start. You must also provide paths to IMGT V and D allele FASTA files when calculating trimming metrics. Help for each script can be accessed using script_name.py -h.
These scripts apply very limited pre-processing to the provided sequences to allow for flexibility and will only drop sequences that contain missing values for required columns. The user may wish to filter to only productive sequences in advance. Some scripts allow the specification of a group column. In these cases this column will be used to group sequences by each unique level of this column and the resulting metrics will be averaged. Otherwise metrics are calculated from the provided file as a whole, treating every sequence equally.
Details of the input files and the scripts that can be used to process them are included below. These scripts do require the installation of pandas (tested with v1.3.5)
IMGT alleles
Germline VDJ alleles in FASTA format with IMGT headers. Only alleles that are marked as F (functional) or ORF (open reading frame) will be used for sequence generation:
- IMGT_human_IGHV.fasta
- IMGT_human_IGHD.fasta
- IMGT_human_IGHJ.fasta
AIRRSHIP includes the IMGT dataset as of 18.02.2022. If an updated dataset is used, or the user wishes to use an alternate database, all other files must contain information to support all the genes present in the new dataset (i.e. if IMGT adds new genes or gene families, and you try to substitute in these files, AIRRSHIP may fail as the currently included distributions do not account for them). AIRRSHIP currently only supports alleles with names in the IMGT format,
See Non-human Species for allele files for information on simulating sequences from species other than humans.
Note
To avoid confusion when benchmarking alignments, AIRRSHIP ignores the IMGT allele IGHV3-30-3*03 as it has the same sequence as allele IGHV3-30*04. AIRRSHIP will also not utilise V or J alleles that do not have the expected junctional anchors.
VDJ usage
VDJ gene and family usage, giving the proportion of sequences to include each gene or family:
- IGHV_usage.csv
- IGHV_usage_gene.csv
- IGHD_usage.csv
- IGHD_usage_gene.csv
- IGHJ_usage.csv
- IGHJ_usage_gene.csv
Example:
| v_family | prop |
|---|---|
| IGHV1 | 0.210282513 |
| IGHV1/OR15 | 0.000783292 |
| ... | ... |
Scripts:
create_vdj_files.py [-h] -i INPUT_FILE [--group GROUP]
-i INPUT_FILE, --input_file INPUT_FILE
--group GROUP Name of column containing dataset metadata to group by.
Required columns: v_call, d_call, j_call
Trimming
Distributions of number of nucleotides to be removed from gene ends at the junctions, per gene family:
- V_family_trimming_proportions.csv
- D_5_family_trimming_proportions.csv
- D_3_family_trimming_proportions.csv
- J_family_trimming_proportions.csv
Example:
| v_family | v_3p_del | proportions |
|---|---|---|
| IGHV1 | 0 | 0.406392257 |
| IGHV1 | 1 | 0.290851348 |
| ... | ... | ... |
Scripts:
create_trimming_files.py [-h] -i INPUT_FILE [--imgt_v IMGT_V] [--imgt_d IMGT_D]
-i INPUT_FILE, --input_file INPUT_FILE
--imgt_v IMGT_V Path to IMGT V allele file.
--imgt_d IMGT_D Path to IMGT D allele file.
Required columns: v_call, d_call, v_germline_end, j_germline_start, d_germline_start, d_sequence_end and d_sequence_start.
NP additions
Proportion of NP regions starting with each nucleotide base:
- np1_first_base_probs.csv
- np2_first_base_probs.csv
Example:
| proportion | |
|---|---|
| A | 0.111702543 |
| C | 0.246122379 |
| G | 0.284580985 |
| T | 0.357594093 |
Proportion of NP regions of each length:
- np1_lengths_proportions.csv
- np2_lengths_proportions.csv
Example:
| np1_length | prop |
|---|---|
| 0 | 0.052651438 |
| 1 | 0.040751106 |
| ... | ... |
Transition probabilities for each position in NP region (i.e. the likelihood of the next base being A,C,G or T when the current base is A,C,G or T). For both mutated and unmutated sequences:
- np1_transition_probs_per_position_igdm.csv
- np1_transition_probs_per_position_igag.csv
- np2_transition_probs_per_position_igdm.csv
- np2_transition_probs_per_position_igag.csv
Example:
| Length | Base | A | C | G | T |
|---|---|---|---|---|---|
| 0 | T | 0.169400291 | 0.390251379 | 0.232798504 | 0.207549826 |
| 0 | A | 0.24858057 | 0.238914924 | 0.34526599 | 0.167238516 |
| ... | ... | ... | ... | ... | ... |
Scripts:
create_np_lengths_files.py [-h] -i INPUT_FILE [--group GROUP]
-i INPUT_FILE, --input_file INPUT_FILE
--group GROUP Name of column containing dataset metadata to group by.
Required columns: np1_length, np2_length
create_np_first_bases.py [-h] -i INPUT_FILE
-i INPUT_FILE, --input_file INPUT_FILE
Required columns: np1_length,np2_length,sequence,v_sequence_end,d_sequence_start and j_sequence_start
create_np_transitions_files.py [-h] -i INPUT_FILE
-i INPUT_FILE, --input_file INPUT_FILE
Required columns: np1_length,np2_length,sequence,v_sequence_end,d_sequence_start and j_sequence_start
When producing NP transition matrices, only a single file for each NP region is produced. AIRRSHIP requires two, np[1|2]_transition_probs_per_position_igdm.csv and np[1|2]_transition_probs_per_position_igag.csv, to account for differences in inserted nucleotides following SHM. Therefore this script must be run once on unmutated sequences and once on mutated sequences and the files manually renamed. If required, the same file with different names can be used although this may affect insertion accuracy.
Somatic Hypermutation
Rates of mutation frequency per sequence, per V family:
- mut_freq_per_seq_per_family.csv
Example:
| v_family | mut_freq | proportion |
|---|---|---|
| IGHV1 | 0 | 0.005959742 |
| IGHV1 | 0.1 | 0.003065621 |
| IGHV1 | 0.111111111 | 0.002811285 |
| ... | ... | ... |
Per base mutation rates for the centre base of each unique 5mer (the likelihood of the centre base being one of A,C,G,T for that kmer when pulled from mutated sequences). For each IMGT region and for the CDR and FWR regions in aggregate:
- cdr_kmer_base_usage.csv
- cdr1_kmer_base_usage.csv
- cdr2_kmer_base_usage.csv
- cdr3_kmer_base_usage.csv
- fwr_kmer_base_usage.csv
- fwr1_kmer_base_usage.csv
- fwr2_kmer_base_usage.csv
- fwr3_kmer_base_usage.csv
- fwr4_kmer_base_usage.csv
Example:
| kmer | A | C | G | T |
|---|---|---|---|---|
| GTGCA | 0.054353444 | 0.001915083 | 0.927897253 | 0.015834221 |
| TGCAC | 0.001906821 | 0.901529425 | 0.017524232 | 0.079039522 |
| GCACA | 0.847932735 | 0.043780581 | 0.074638153 | 0.033648531 |
| ... | ... | ... | ... | ... |
Scripts:
create_shm_files.py [-h] -i INPUT_FILE
-i INPUT_FILE, --input_file INPUT_FILE
Required columns: v_call, germline_alignment, sequence_alignment, fwr1, fwr2, fwr3, cdr1, cdr2, cdr3
Experimental data used
The input files included with AIRRSHIP were generated using repertoire data from the following public datasets:
- Cowan, G et al., unpublished data.
- Ghraichy,M. et al. (2020) Maturation of the Human Immunoglobulin Heavy Chain Repertoire With Age. Front. Immunol., 11, 1734.
- Preprocessed data downloaded from https://zenodo.org/record/3585046#.Y2upy4LP2JF.
- Only individuals aged > 9 years included.
- Gidoni,M. et al. (2019) Mosaic deletion patterns of the human antibody heavy chain gene locus shown by Bayesian haplotyping. Nat. Commun., 10, 628.
- Raw sequence data downloaded from the ENA, accession PRJEB26509.
- Healthy controls only included.
- Waltari,E. et al. (2018) 5′ Rapid Amplification of cDNA Ends and Illumina MiSeq Reveals B Cell Receptor Features in Healthy Adults, Adults With Chronic HIV-1 Infection, Cord Blood, and Humanized Mice. Front. Immunol., 9, 628.
- Raw sequence data downloaded from the SRA, accession PRJNA393446.
- Healthy adults only included.
- Yang,X. et al. (2021) Large-scale analysis of 2,152 Ig-seq datasets reveals key features of B cell biology and the antibody repertoire. Cell Rep., 35, 109110.
- Raw sequence data downloaded from the SRA, accession PRJNA564936.
Sequences were processed using pRESTO [1] and Change-O [2] from the Immcantation pipeline, with VDJ assignment carried out using IgBLAST [3] v1.18.0.
[1] Vander Heiden, J. A. et al. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932 (2014).
[2] Gupta, N. T. et al. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358 (2015).
[3] Ye, J., Ma, N., Madden, T. L. & Ostell, J. M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 41, W34–W40 (2013).
Simulating Non-human Species
AIRRSHIP has not specifically been designed to allow the simulation of sequences from non-human sequences and does not come with in-built reference files for other species. However, if the species of interest has a recombination process that follows the same steps as are present in humans, it should be possible for a user to simulate from a reference directory containing files generated from this species.
The --species flag must be used to find the VDJ allele files if simulating non-human sequences. File names are expected to be in the format imgt_[species]_IGH[V|D|J].fasta.
AIRRSHIP is primarily intended for simulation of human sequences and has been tested on this basis. If you are simulating non-human sequences, ensure the output looks as you would expect. Any unusual behaviour or errors by can be reported by raising an issue on Github.
Mouse Reference Datasets
We have tested this premise by simulating sequences from C57BL/6 mice and reference files are available at the AIRRSHIP Github. These reference files were produced from the datasets detailed below. The germline alleles included are those that are present in both the IMGT mouse reference files and the OGRDB C57BL/6 reference germline set [1,2]. The number of individuals and sequences is considerably smaller than that used to generate the human references. We have also not fully optimised AIRRSHIP for non-human data, and as a result the simulations are slightly less realistic than for human sequences, especially across the junction region.
To simulate using these files, the following command would be used after the c57bl6 reference directory had been downloaded:
airrship -o c57bl6_repertoire \
--datadir path/to/c57bl6_reference \
--species mouse
Experimental data used
- Collins, A.M. et al. (2015) The mouse antibody heavy chain repertoire is germline-focused and highly variable between inbred strains. Philos. Trans. R. Soc. B Biol. Sci., 370.
- Raw sequence data downloaded from ENA accession PRJEB8745
- Only C57BL/6 samples used
- Corcoran, M.M. et al. (2016) Production of individualized V gene databases reveals high levels of immunoglobulin genetic diversity. Nat. Commun. 2016 71, 7, 1–14.
- Raw sequence data downloaded from ENA accession PRJEB15295
- C57Bl/6 M3 sample used
- Greiff, V. et al. (2017) Systems Analysis Reveals High Genetic and Antigen-Driven Predetermination of Antibody Repertoires throughout B Cell Development. Cell Rep., 19, 1467–1478.
- Raw sequence data downloaded from ArrayExpress accession E-MTAB-5349
- Naive splenocyte samples from uninfected C57BL/6 used
- Kräutler, N.J. et al. (2020) Quantitative and Qualitative Analysis of Humoral Immunity Reveals Continued and Personalized Evolution in Chronic Viral Infection. Cell Rep., 30, 997-1012.e6.
- Raw sequence data downloaded from ArrayExpress accession E-MTAB-8585
- Mouat, I. et. al., unpublished data
- Splenocytes from eight control C57BL/6 mice
[1] Lees, W. et al. OGRDB: a reference database of inferred immune receptor genes. Nucleic Acids Res. 48, D964–D970 (2020).
[2] Jackson, K. J. L. et al. A BALB/c IGHV Reference Set, Defined by Haplotype Analysis of Long-Read VDJ-C Sequences From F1 (BALB/c x C57BL/6) Mice. Front. Immunol. 13, 2490 (2022).
Release notes
We recommend using the latest available AIRRSHIP release. If you have already installed an earlier version then you may have to upgrade your installed version using pip.
pip install airrship -U
v0.1.0
Initial release.
v0.1.1
Bug fix: the --shm_random option no longer produces an error.
v0.1.2
Adjustments to ensure that proportional VDJ usage is accurately recreated from reference files.
v0.1.3
Added option for including non-productive sequences in output. Added --shm_multiplier option to control mutation rates.
v0.1.4
Yanked. Issue with Python 3.7 compatibility.
v0.1.5
Included --species option to make simulating non-human species using user specified data easier.
Warning
Default values for --het have changed to 0 0 0. This differs from previous releases where the default behaviour was to use the greatest proportion of heterozygous positions possible in the inbuilt data.